
By Dr Alex Tan Weixian, Consultant, Department of Cardiology
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiomyopathy, affecting 1 in 500. The condition is defined by left ventricular hypertrophy (LVH) of ≥15mm or ≥13mm in cases with a family history of HCM, in the absence of another disease process that can cause LVH, such as hypertension, valvular heart disease, or infiltrative heart disease.
HCM is caused by mutations of genes coding for sarcomeric proteins, which are proteins involved in heart muscle contraction. The condition is transmitted in an autosomal dominant pattern, meaning children of an affected parent have a 50% chance of inheriting the mutation. When examining patients with LVH, one should consider the mimickers of LVH, such as cardiac amyloidosis, Fabry's Disease, and Danon's Disease. Identifying these conditions is crucial as they have different complications and treatment approaches from HCM.
The Path to Identifying Hypertrophic Cardiomyopathy
Diagnosis begins with a clinical suspicion based on symptoms, family history, and at times, a cardiac murmur. Symptoms of HCM may initially appear vague and non-specific, including breathlessness, chest pain, palpitations, light-headedness, and fainting.
The next step in investigation will be an electrocardiogram (ECG) which may show LVH changes. However, the absence of LVH changes on ECG does not rule out HCM. Cardiac imaging is therefore important in supporting the diagnosis. Echocardiography remains the cornerstone in diagnosing LVH and HCM. It also helps with assessment of left ventricular ejection function (LVEF) and any complications associated with HCM. Cardiac MRI may be performed when the echocardiogram is non-diagnostic or to assess for mimickers of LVH and complications of HCM.
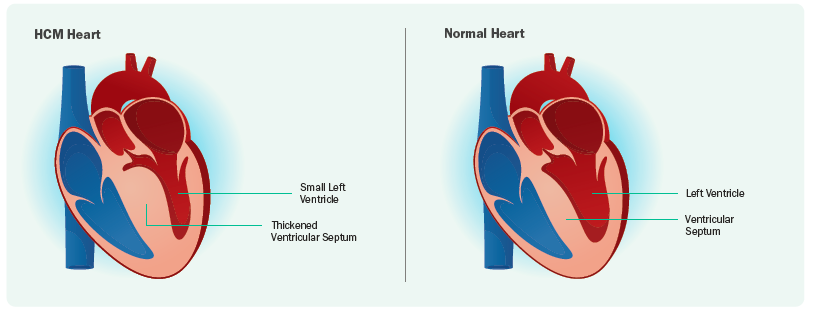
The Clinical Spectrum of Hypertrophic Cardiomyopathy
HCM can be divided into obstructive and non-obstructive. Obstructive HCM occurs when there is a combination of systolic anterior motion (SAM) of the mitral valve leaflet, where the mitral valve moves abnormally forward during heart contraction, combined with a thickened left ventricle septum. This leads to SAM-septal contact and left ventricular outflow tract (LVOT) obstruction, making it difficult for blood to exit the heart (left ventricle) to the body. Obstruction can be measured on an echocardiogram and is considered to be present if the peak LVOT gradient is ≥30 mm Hg. Symptoms typically worsen with exertion and certain conditions such as dehydration, hot weather, large meals, and sudden posture changes (sitting to standing). Patients may feel easily out of breath or light-headed, and in severe cases, may experience fainting (syncope). Up to approximately two-thirds of HCM patients may have LVOT obstruction.
LVOT obstruction can be diagnosed via echocardiogram. Sometimes, when the resting echocardiogram does not show LVOT obstruction, an exercise (treadmill or bicycle) stress echocardiogram is performed to identify latent LVOT obstruction. Diagnosing LVOT obstruction early is important as symptoms can be improved through lifestyle measures and treatment.
Other complications of HCM include:
- Atrial fibrillation (AF), which increases the risk of thromboembolism and stroke. Regular clinical assessments and ambulatory ECG monitoring (eg. Holter) can help detect AF, which may be silent. If diagnosed, oral anticoagulation should be prescribed.
- Microvascular ischaemia can lead to chest pain in the absence of obstructive coronary artery disease. This condition can be treated effectively with medications, such as beta-blockers, verapamil, diltiazem and ranolazine.
- Heart failure, occurring as either systolic dysfunction or diastolic dysfunction. Systolic dysfunction in HCM occurs when LVEF is <50% and affects <5% of HCM patients. Treatment includes heart failure medications and fluid status monitoring.
- Risk of sudden cardiac death due to malignant ventricular arrhythmias, which has led to development of risk stratification methods to assess the need for an implantable cardioverter defibrillator (ICD).
A Comprehensive Approach to Treatment
The treatment of obstructive HCM is symptom-guided, encompassing lifestyle strategies, medications and invasive therapies. Simple lifestyle measures can significantly improve symptoms.
Medications play a key role in alleviating symptoms of LVOT obstruction. These include non-vasodilating beta-blockers (eg. Bisoprolol) or non-dihydropyridine calcium channel blockers (eg. verapamil and diltiazem), which help to slow the heart rate and reduce contractility. If symptoms persist, disopyramide, through its negative inotropic effect (reduces heart muscle contractility) can also be added.
If symptoms remain significant, invasive strategies of either alcohol septal ablation or surgical myectomy can be employed to reduce the septal wall thickness and alleviate LVOT obstructions. While these treatment options are effective, they are limited to treating the downstream effects of HCM. In recent years, a new drug, Mavacamten, was designed to target the actual disease process of HCM and has been proven to effectively reduce LVOT obstruction and related symptoms.
Family Screening and Genetic Testing
When HCM is diagnosed, family screening is recommended. First-degree relatives should undergo clinical screening with ECG and echocardiogram for HCM. Even if the initial screening is negative, repeated screening should be considered until the relative is around 60 years of age, after which the development of HCM becomes less likely.
Genetic testing, together with appropriate counselling, is also recommended. This is usually done via a blood test that will be checked for abnormal genes that can cause LVH. The role of genetic testing is to confirm the diagnosis of HCM - an abnormal (pathogenic) gene causing HCM may be found in about 30-60% of cases. In addition, genetic testing can help rule out mimickers of HCM. Lastly, it also allows cascade screening of relatives, that is, systematic screening of family members based on identified genetic mutations, if an abnormal gene causing HCM is found.
In conclusion, with increased awareness and earlier diagnosis of HCM, symptoms and complications can be effectively managed with an expanding range of medications and procedures. Long-term follow-up is essential to monitor for new symptoms or complications. Early diagnosis, starting with clinical suspicion and appropriate investigations including ECG and echocardiogram, followed by cardiologist's review, remains key to optimal care.
REFERENCES
1. Ommen SR, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2024 Jun 4;149(23):e1239-e1311.
2. Olivotto Iacopo, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet, Volume 396, Issue 10253, 759 - 769
3. Maron MS, et al.Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006 Nov 21;114(21):2232-9.
4. Girolami F, et aI. Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions. J Clin Med. 2023 Mar 24;12(7):2489.
This article is from Murmurs Issue 49. Click here to read other articles or issues.
