











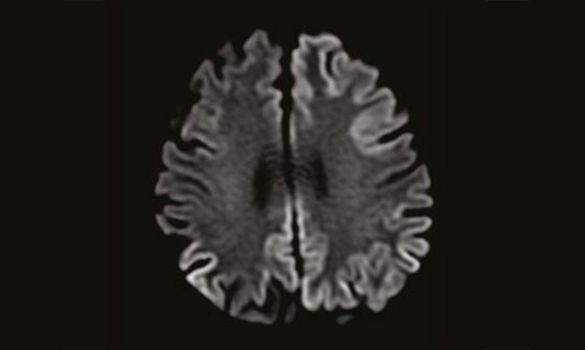
MRI showing Cortical Restricted Diffusion
CASE 1
A 70-year-old Chinese man with no past medical history of note presented with a 2-week history of cognitive impairment.
He had problems with short-term recall, being unable to remember a list of groceries to buy, misplacing items and forgetting to turn off the tap. He also had difficulty turning on his computer. In terms of language, he was noted to have word-finding difficulty and a poverty of speech.
On clinical examination, he was oriented to the time, place and person, but not the day nor date. He was unable to perform the 3-item recall, and was noted to have perseveration of speech and dysphasia. Limb examination was remarkable for bilateral lower limb rigidity.
Blood investigations were unremarkable for any metabolic, autoimmune or infective causes. An MRI of the brain showed cortical diffusion restriction, involving both the temporal and parietal lobes bilaterally, as well as the cingulate gyri and left frontal lobe. [Refer to Image 1]
An electroencephalogram (EEG) showed periodic sharp wave complexes in the left hemisphere, and cerebrospinal fluid (CSF) analysis was normal. A diagnosis of rapidly progressive dementia, secondary to sporadic Creutzfeldt-Jakob disease (CJD), was made.
The patient’s symptoms progressed rapidly. He developed myoclonic jerks and was treated symptomatically with clonazepam and levetiracetam. A month after the onset of symptoms, he had become fully dependent and was in an akinetic mute state. He was subsequently transferred to the inpatient hospice care.
CASE 2
A 67-year-old Malay housewife, who was previously well, was admitted to the hospital for a 5-minute episode of transient unresponsiveness during a religious class. Extensive investigations with an MRI of the brain, EEG and telemetry monitoring for cardiac arrhythmias were negative. She was discharged with an outpatient Neurology referral.
Over the next 3 months, the patient complained of shortterm memory loss. For example, she had no memory of having gone to her grandson’s primary school, despite being there several times during that period. Her husband noted that she became progressively more anxious, irritable and socially withdrawn. In addition, she also complained of intermittent, brief episodes of chills associated with goose bumps and abnormal epigastric sensation, up to 20 times a day.
An outpatient cognitive testing revealed the evidence of reduced attention and executive dysfunction. A repeat EEG showed epileptiform activity in the left frontal and temporal lobes, suggesting that her episodes of “chills” were due to focal seizures.
A repeat MRI scan of the brain was normal, and a CSF analysis was unremarkable. The initial blood investigations were also normal.
A presumptive diagnosis of possible autoimmune limbic encephalitis was made on the basis of her cognitive decline, personality change and focal seizures. The testing of serum for autoimmune encephalitis antibodies subsequently came back positive for the anti-LGI1 receptor antibody, thus confirming the diagnosis.
She was treated with high dosage intravenous methylprednisolone, followed by a tapering dose of oral corticosteroids. Rapid improvement in her cognitive function and seizure control ensued.
Dementia is defined as a decline in cognition associated with functional impairment. In Rapidly Progressive Dementia (RPD), deterioration from the symptom onset to a diagnosis of dementia progresses at a rate faster than that expected for typical dementia. Though a time frame of 2 years is sometimes used, most cases of RPD develop sub-acutely, from weeks to months 1.
Table 1 Causes of Rapidly Progressive Dementia
| Causes of Rapidly Progressive Dementia |
|
Prion disease e.g. Creutzfeldt-Jakob disease (CJD) |
|
Non-prion Neurodegenerative disease e.g. Frontotemporal Dementia, Corticobasal Syndrome, Alzheimer disease |
|
Immune-mediated/Paraneoplastic e.g. Limbic Encephalitis |
|
Vascular e.g. Ischaemic/Haemorrhagic Stroke, Subdural Haemorrhage, Vascular Dementia, CNS Vasculitis |
|
Neoplasm e.g. Metastases, CNS Lymphoma |
|
Infection e.g. HSV, HIV, Progressive Multifocal Leukoencephalopathy (PML), Syphilis, Whipple disease |
|
Toxic-metabolic e.g. Electrolyte and Endocrine abnormalities, Illicit drug use |
Most patients with RPD present in the 6th or 7th decade of life, though patients as young as 12-years-old have been reported. This is because RPD is essentially a clinical syndrome, which encompasses many varied etiologies [Refer to Table 1].
In the adult population, most are due to irreversible neurodegenerative diseases – usually rapidly progressive forms of more common dementias, such as frontotemporal dementia, corticobasal syndrome, vascular dementia and rarely, Alzheimer disease and dementia with Lewy bodies 2,3.
The other important group of neurodegenerative diseases are those due to abnormal prion protein deposition, such as Creutzfeldt-Jakob disease (CJD). A typical patient is highlighted within Case 1. Patients with CJD present with global cognitive decline and other neurological features, including cerebellar ataxia, visual disturbances, parkinsonism and myoclonus.
Diagnosis is made by a combination of clinical, radiological and EEG criteria. A brain biopsy is rarely necessary. The treatment is palliative, and the disease is invariably fatal with a median survival of 6 - 11 months.
The incidence of CJD is one per million of the population per year. At the National Neuroscience Institute at the Tan Tock Seng Hospital Campus, we see an average of 3 cases per year, with 29 cases diagnosed from 2006 to 2016 4.
More importantly, however, between 17% to 23% of RPD patients have potentially treatable etiologies 1, which include immune-mediated, paraneoplastic, neoplastic, infective and toxic-metabolic conditions [Refer to Table 1].
In these patients, the reversibility of dementia improves with early diagnosis and treatment. Hence, it is incumbent on the evaluating physician or neurologist to perform a comprehensive evaluation to identify a treatable cause of RPD.
SYMPTOMS AND INITIAL EVALUATION
Perhaps the most crucial part of the initial evaluation of a patient with RPD is to obtain the clinical history from a reliable historian and, wherever possible, from multiple corroborative sources 1.
The first step is to exclude the presence of delirium, often due to a reversible systemic cause, which can cause the rapid decline of pre-existing cognitive impairment or dementia 2.
Secondly, some patients who present with so-called RPD may, in actual fact, already have long-standing undiagnosed dementia. Subtle cognitive decline may have progressed insidiously for a few years, but patients present only when the memory loss is significant enough to come to their family members’ attention - thus mimicking an RPD. This is especially so for non-Alzheimer dementias, that tend to cause executive dysfunction and personality changes, with the relative sparing of short-term memory. Hence, a corroborative history from multiple sources is important.
The next step is to establish the progression of symptoms. An abrupt onset or stepwise progression is the hallmark of a vascular etiology, while an acute presentation over days may indicate an infective or toxic-metabolic cause.
In RPD, the initial presenting symptoms and signs are also useful for neurolocalisation and diagnosis. This requires a detailed evaluation, not only of the classical symptoms of dementia (amnesia, agnosia, aphasia, apraxia, agnosia, executive dysfunction), but also other features that localise to various affected regions of the brain. Therefore, a detailed neurological examination is compulsory to identify signs of cerebellar, extrapyramidal or brainstem dysfunction 1.
In Case 2, our patient presented with short-term memory loss, focal seizures with epigastric aura and a personality change, all of which localise to the limbic system. This led to the clinical suspicion and eventual diagnosis of autoimmune limbic encephalitis, a treatable cause of RPD 5.
The presence of systemic symptoms and the patient’s medical and drug history also help to narrow down the differential diagnoses. Common drugs that may affect cognition, particularly in elderly patients, include anticholinergics and benzodiazepines. In a patient with risk factors for sexually transmitted diseases, HIV and syphilis need to be considered; while a patient with anorexia and cachexia may be harbouring a malignancy.
WHAT IS THE ROLE OF THE GENERAL PRACTITIONER (GP)?
A GP who encounters a patient who is suspected of RPD should not hesitate to refer him or her to a specialist Neurology clinic for an early evaluation. Screening of the renal and liver function, electrolytes (especially of sodium and calcium), thyroid function, vitamin B12 and folate levels – and where relevant, HIV testing – can also be performed in the primary care setting.
Abrupt or rapid progression of symptoms over days should prompt a referral to the Emergency department, not only for the exclusion of a life-threatening cause, but also for prompt evaluation and treatment.
WHAT ARE THE LIKELY TREATMENT OPTIONS BY THE SPECIALIST?
In the Neurology clinic, a full evaluation of the patient’s cognition and an MRI of the brain are obligatory. Further ancillary investigations, such as an EEG, CSF analysis and more specialised blood investigations, are often necessary. Depending on the clinical context, systemic evaluation with CT or PET imaging for occult imaging may also be performed.
Treatment options depend on the etiology of the RPD, ranging from symptomatic (in the context of neurodegenerative diseases) to potentially reversible (in the setting of infections, metabolic disorders, autoimmune conditions and neoplasms).
WHEN WILL THE PATIENT BE REFERRED BACK TO THE GP?
Patients with treatable conditions (for example, hypothyroidism) may be referred back to their GP when specialist titration of treatment is no longer required.
Some patients with neurodegenerative dementias, such as Alzheimer or vascular dementia, whose symptoms are well-controlled with medications such as cognitive enhancers, antidepressants and/or antipsychotics, can also be followed up by their GP. Patients with limited life expectancy may either be transferred to inpatient hospice care or discharged to the care of a primary care physician in the home care setting.
GPs can call for appointments through the GP Appointment Hotline at 6357 7095 or scan the QR code for more information.
By: Dr Chiew Hui Jin, Associate Consultant, Department of Neurology, National Neuroscience Institute (NNI)
Dr Chiew Hui Jin is an Associate Consultant at the Department of Neurology, National Neuroscience Institute. His interests are in Cognitive Neurology and in Medical Education.
REFERENCES
1. Geschwind MD. Rapidly progressive dementia. Continuum (Minneap Minn). 2016 Apr;22(2 Dementia):510-37. 2. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol 2008;64(1):97-108.
3. Mead S. P Rudge. CJD Mimics and Chameleons. Pract Neurol. 2017 Apr;17(2):113-121.
4. Clinical and radiological characteristics in Creutzfeldt-Jakob Disease from a tertiary referral centre in Singapore. Chiew HJ. Kandiah N. Ng AS. Poster presentation.
5. Graus F, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391–404.

Stay Healthy the Easy Way
Get trusted health advice, offers and more.

Get the Health Buddy App



Follow HealthXchange

